5 Database schema
We initially carry over the database schema from pyani, as illustrated in Figure 5.1. There are four key tables and three linker tables, discussed below.
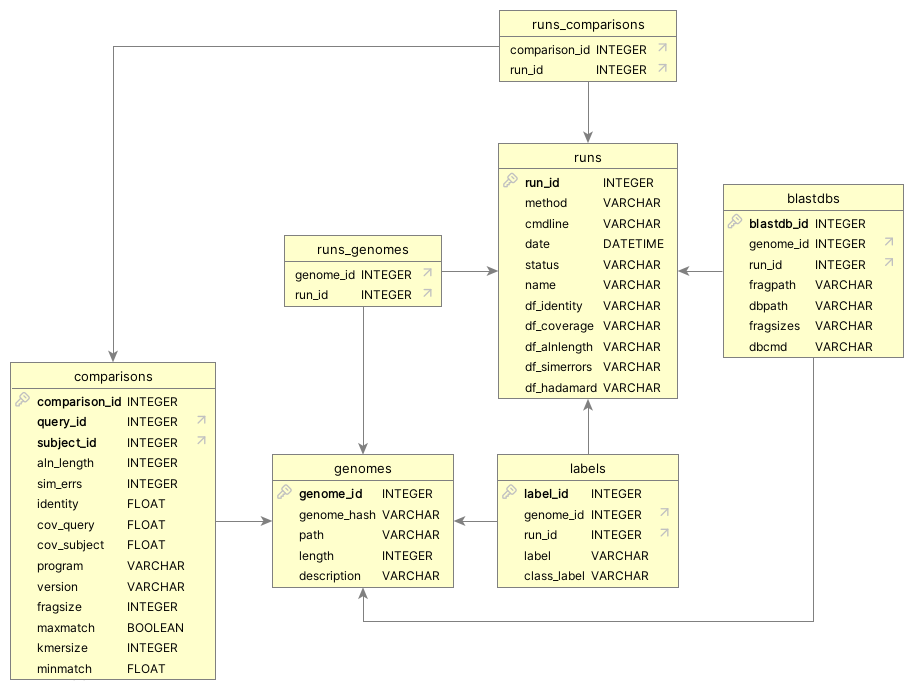
pyani database schema
An empty database with this schema can be found at the link below.
5.1 Tables
The central concept of the database is the comparison, which describes the outcome of a pairwise comparison between two genomes. Each comparison is performed as part of a run - a batch of one or more comparisons carried out “at the same time” (or, at least, under the same command-line instruction).
Each genome can carry one or more labels - metadata describing the genome. A single genome can have different labels in each run it is associated with.
For BLAST-based comparisons, such as anib, a set of fragmented sequences may be created, and a BLAST database generated. Details of these are stored in the blastdbs table.
As a single run may comprise many comparisons between many genomes, two linker tables are required: runs_comparisons and runs_genomes
The schema is not set in stone. As new analyses are included in pyani-plus or we want to record additional data, the schema may change. We will manage versioning of the schema using Alembic.
5.1.1 comparisons
The comparisons table is a superset of the data we would expect to receive relating to a single pairwise genome comparison. It includes fields that are general to all comparisons (e.g. reporting of identity and coverage scores, the query and subject genome IDs, details of how the comparison was performed), and those that are relevant to some methods but not others. See Figure 5.1 for how the comparisons table relates to other tables in the schema.
The SQL table definition is given below.
sqlite> .schema comparisons
CREATE TABLE comparisons (
comparison_id INTEGER NOT NULL,
query_id INTEGER NOT NULL,
subject_id INTEGER NOT NULL,
aln_length INTEGER,
sim_errs INTEGER,
identity FLOAT,
cov_query FLOAT,
cov_subject FLOAT,
program VARCHAR,
version VARCHAR,
fragsize INTEGER,
maxmatch BOOLEAN,
kmersize INTEGER,
minmatch FLOAT,
PRIMARY KEY (comparison_id),
UNIQUE (query_id, subject_id, program, version, fragsize, maxmatch, kmersize, minmatch),
FOREIGN KEY(query_id) REFERENCES genomes (genome_id),
FOREIGN KEY(subject_id) REFERENCES genomes (genome_id)
);comparison_id: the primary key, an integer that increments with each additionalcomparisonrowquery_id: foreign key identifying the query genome in the comparison (genomestable)subject_id: foreign key identifying the subject/reference genome in the comparison (genomestable)aln_length: the length of the alignment, on the query sequencesim_errs: the count of similarity errors, relevant toMUMmercomparisons onlyidentity: the (query-wise) percentage identity of the alignment (typically the count of identical bases in the alignment, divided by the non-overlapping alignment length)cov_query: the percentage proportion of the query genome involved in the (non-overlapping) alignmentcov_subject: the percentage proportion of the query genome involved in the (non-overlapping) alignmentprogram: the name of the program used to conduct the alignmentversion: the version number of the program used to conduct the alignmentfragsize: fragment length used in BLAST-based andfastANIcomparisonsmaxmatch: boolean value indicating whether the--maxmatchoption was used forMUMmer-based analysiskmersize: the k-mer length used in k-mer-based comparisonsminmatch: infastANIcomparisons, the minimum matching fraction of the genome required to accept the comparison output
In general we want to avoid repeating comparisons unless there is a material change to the comparison. Hence we have a UNIQUE constraint on the combination of: query genome; subject genome; program used; program version; parameter settings (fragment size, maxmatch setting, k-mer size, and minimum aligning fraction).
In transitioning to pyani-plus the fields in this table may be changed to accommodate new analyses and parameter choices, or for other reasons to improve the code.
It may make sense, in the transition to pyani-plus to move the information that is specific to a run and not to a comparison to the runs table.
If we want to enable runs that apply more than one method or parameter set, the current schema is repetitious, but the simplest route to accommodating that kind of operation. If we don’t move to that kind of operation, it makes sense to refactor out the run-level information.
5.1.2 genomes
Genomes are the FASTA file inputs to each comparison. A genome is considered unique if its FASTA header and sequence combination are unique. This can be determined by taking a suitable hash of the genome sequence file.
Changing the filename or file extension of a genome does not alter the genome.
In pyani we used the MD5 hash:
fname = Path(fname) # ensure we have a Path object
hash_md5 = hashlib.md5() # nosec
try:
with fname.open("rb") as fhandle:
for chunk in iter(lambda: fhandle.read(65536), b""):
hash_md5.update(chunk)We want to avoid repeating analyses or extending the database size unnecessarily wherever possible, so genome hashes are checked before inclusion of genomes into the table, and before scheduling a comparison to run.
The SQL table definition is given below.
sqlite> .schema genomes
CREATE TABLE genomes (
genome_id INTEGER NOT NULL,
genome_hash VARCHAR,
path VARCHAR,
length INTEGER,
description VARCHAR,
PRIMARY KEY (genome_id),
UNIQUE (genome_hash)
);genome_id: the primary key, an integer that increments with each additionalgenomerowgenome_hash: a hash of the contents of the genome sequence FASTA filepath: path to the location of the file (this may not be the only location corresponding to the genome file)length: genome sequence length (sum of all sequence lengths in the file)description: description string from the FASTA header
5.1.3 runs
A run in this context is a single invocation of pyani-plus. This will typically involve an input directory containing multiple genome files, and specification of a single analysis mode, implying a specific sequence comparison tool and associated parameters. We record relevant information at the run level, such as the command-line used to execute pyani-plus, the date, and an optionally meaningful name for the user to recover results.
The SQL table definition is given below.
sqlite> .schema runs
CREATE TABLE runs (
run_id INTEGER NOT NULL,
method VARCHAR,
cmdline VARCHAR,
date DATETIME,
status VARCHAR,
name VARCHAR,
df_identity VARCHAR,
df_coverage VARCHAR,
df_alnlength VARCHAR,
df_simerrors VARCHAR,
df_hadamard VARCHAR,
PRIMARY KEY (run_id)
);run_id: the primary key, an integer that increments with each additional `run`` rowmethod: the name of thepyani-plusmethod that was called.cmdline: the command-line that was used to invokepyani-plusdate: the date/time that the command was runstatus: current status of the run (e.g.started,complete,errored)name: user-provided name for the run
It may make sense, in the transition to pyani-plus to move the information that is specific to a run and not to a comparison to the runs table, rather than under comparisons.
If we want to enable runs that apply more than one method or parameter set, the current schema is repetitious, but the simplest route to accommodating that kind of operation. If we don’t move to that kind of operation, it makes sense to refactor out the run-level information.
5.1.4 labels
The labels table is intended to provide a way to label each genome with metadata that can be used for downstream processing of run output, such as membership of a taxonomic group, possession of a phenotype, and so on. As the metadata for a genome may vary if it participates in more than one run, this is effectively a decorated linker table between runs and genomes.
The SQL table definition is given below.
sqlite> .schema labels
CREATE TABLE labels (
label_id INTEGER NOT NULL,
genome_id INTEGER,
run_id INTEGER,
label VARCHAR,
class_label VARCHAR,
PRIMARY KEY (label_id),
FOREIGN KEY(genome_id) REFERENCES genomes (genome_id),
FOREIGN KEY(run_id) REFERENCES runs (run_id)
);labels_id: the primary key, an integer that increments with each additional `run`` rowgenome_id: the genome to which the label appliesrun_id: the run for which the label applieslabel: the text of the metadataclass_label: the text of the class metadata
Two slots are provided: label and class_label. This is quite inflexible in comparison to what we might like, and was written to match the original implementation in average_nucleotide_identity.py.
We should make this table, and the way we access it for label information, more general in pyani-plus. One approach might be to modify the table to:
CREATE TABLE labels (
label_id INTEGER NOT NULL,
genome_id INTEGER,
run_id INTEGER,
label VARCHAR,
label_type VARCHAR,
PRIMARY KEY (label_id),
FOREIGN KEY(genome_id) REFERENCES genomes (genome_id),
FOREIGN KEY(run_id) REFERENCES runs (run_id)
);Allowing for multiple labels per genome, per run, with a label_type field that uses a controlled vocabulary to indicate the type of metadata that is encoded in the label.